Chemistry in Silico
Research in the Parish group focuses on understanding the dynamical behavior of interesting molecular systems. The tools of quantum mechanics, conformational searching and free energy simulations are used to answer questions about the structure, energy and dynamics of these systems. We develop new tools as needed and refine and optimize existing tools. Research funding for these projects has been obtained from the NSF, ACS-PRF and the Camille and Henry Dreyfus Foundation.
Polyhedral Oligomeric Silsesquioxanes (POSS): Silsesquioxanes are macromolecules that contain silicon and oxygen in a 1:1.5 atom ratio and refer to random structures, ladder structures, cage structures, and partial cage structures, as illustrated in Figure 1.
 Although first reported in 1946, between that
time and the late 1990s, silsesquioxanes received little attention in the
literature. Interest in these
systems has exploded recently due to their commercial availabilty and their
potential for use in nanoscale devices, however very little is known about
their molecular behavior. Our research
group has been investigating the electronic structure, conformational flexibility
and dynamical behavior of silsesquioxanes and their derivatizes
in order to correlate structure with anticipated nanomolecular function. We are perfoming molecular dynamics simulations
on caged structures to investigate the “breathability” of these systems and
to determine how the electronic structure of the derivatized systems effects
the pore size. We are also using conformational searching methods
to probe the flexibility of partial cage and ladder structures and to determine
the suitability of these systems for use in host-guest chemistry.
Although first reported in 1946, between that
time and the late 1990s, silsesquioxanes received little attention in the
literature. Interest in these
systems has exploded recently due to their commercial availabilty and their
potential for use in nanoscale devices, however very little is known about
their molecular behavior. Our research
group has been investigating the electronic structure, conformational flexibility
and dynamical behavior of silsesquioxanes and their derivatizes
in order to correlate structure with anticipated nanomolecular function. We are perfoming molecular dynamics simulations
on caged structures to investigate the “breathability” of these systems and
to determine how the electronic structure of the derivatized systems effects
the pore size. We are also using conformational searching methods
to probe the flexibility of partial cage and ladder structures and to determine
the suitability of these systems for use in host-guest chemistry.
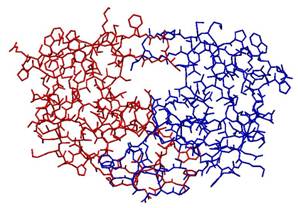 HIV Protease Inhibitor Drugs: The
human immunodeficiency virus (HIV) encodes an aspartyl protease enzyme (HIV-PR)
that cleaves viral polyprotein precursors, allowing the maturation of the
HIV virus that causes the autoimmune deficiency disease (AIDS).
Protease inhibitors occupy the basket-shaped active site, interfering
with the functioning of the enzyme and preventing the maturation of the virus.
Our work uses conformational searching and free energy simulation methods
to study different FDA-approved HIV protease inhibitor drugs such as indinavir,
saquinavir, nelfinavir, amprenavir, lopinavir and ritonavir to determine the
similarities and differences in the way these drugs interact with the protease
active site. For instance, functional
group interactions and conformational flexibility are thought to be important
in the binding of the inhibitor to the protease.
Models have been proposed for specific
inhibitor-protease interactions, however there has not been a systematic comparison
of known inhibitors ref. This
study will address the following questions: What common structural motifs
do active HIV-PR inhibitors share? Are hydrogen bonding patterns the same among
all inhibitors or unique to individual inhibitors? If unique, is this due to flexibility on the
part of the protease? What role do
solvent interactions play? This information
will be used to develop a “prescription” or template for describing molecular
behavior that is necessary in the design of new and more effective protease
inhibitors. We have completed an in-depth
analysis of two recently developed conformational searching methods and we
are confident that Low Mode:
HIV Protease Inhibitor Drugs: The
human immunodeficiency virus (HIV) encodes an aspartyl protease enzyme (HIV-PR)
that cleaves viral polyprotein precursors, allowing the maturation of the
HIV virus that causes the autoimmune deficiency disease (AIDS).
Protease inhibitors occupy the basket-shaped active site, interfering
with the functioning of the enzyme and preventing the maturation of the virus.
Our work uses conformational searching and free energy simulation methods
to study different FDA-approved HIV protease inhibitor drugs such as indinavir,
saquinavir, nelfinavir, amprenavir, lopinavir and ritonavir to determine the
similarities and differences in the way these drugs interact with the protease
active site. For instance, functional
group interactions and conformational flexibility are thought to be important
in the binding of the inhibitor to the protease.
Models have been proposed for specific
inhibitor-protease interactions, however there has not been a systematic comparison
of known inhibitors ref. This
study will address the following questions: What common structural motifs
do active HIV-PR inhibitors share? Are hydrogen bonding patterns the same among
all inhibitors or unique to individual inhibitors? If unique, is this due to flexibility on the
part of the protease? What role do
solvent interactions play? This information
will be used to develop a “prescription” or template for describing molecular
behavior that is necessary in the design of new and more effective protease
inhibitors. We have completed an in-depth
analysis of two recently developed conformational searching methods and we
are confident that Low Mode:
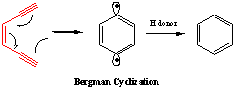 Anticancer Enediyne Warhead Drugs: Naturally
occurring anticancer agents, such as Dynemicin A, contain reactive, electron
rich enediyne moieties shown in red. Under the proper conditions, the enediyne group
undergoes a Bergman cyclization that results in a p-benzyne diradical that
can abstract hydrogen atoms from DNA resulting
Anticancer Enediyne Warhead Drugs: Naturally
occurring anticancer agents, such as Dynemicin A, contain reactive, electron
rich enediyne moieties shown in red. Under the proper conditions, the enediyne group
undergoes a Bergman cyclization that results in a p-benzyne diradical that
can abstract hydrogen atoms from DNA resulting  in cancer cell death. In order to design
anticancer agents or other endiynes of biological interest it is important
to understand the factors that affect the rate of Bergman cyclization. This study will use quantum mechanical and reaction
field theory methods to understand how the rate of Bergman cyclization in
ten-membered enediynes is influenced by various electronic factors. Using appropriate model systems we will investigate
the effects of pH, molecular charge distribution, tautomerism and solvation.
High level ab
initio calculations will be performed on each
of the enediynes prepared in our collaborator’s laboratory (KC Russell, Northern
Kentucky University) in order to evaluate their structure, stability, cyclization
barriers, thermochemistry and singlet-triplet energy differences.
Our theoretical results will be verified by comparison with experimentally
obtained temperature dependent rate constants obtained in the Russell group. Together, these studies will provide molecular
level insight into the competing factors influencing cyclization.
An understanding of how these factors effect the rate of cyclization
will allow the development of more effective anticancer prodrug warheads –
enediynes that cyclize and cause cell death only under certain, controlled
conditions.
in cancer cell death. In order to design
anticancer agents or other endiynes of biological interest it is important
to understand the factors that affect the rate of Bergman cyclization. This study will use quantum mechanical and reaction
field theory methods to understand how the rate of Bergman cyclization in
ten-membered enediynes is influenced by various electronic factors. Using appropriate model systems we will investigate
the effects of pH, molecular charge distribution, tautomerism and solvation.
High level ab
initio calculations will be performed on each
of the enediynes prepared in our collaborator’s laboratory (KC Russell, Northern
Kentucky University) in order to evaluate their structure, stability, cyclization
barriers, thermochemistry and singlet-triplet energy differences.
Our theoretical results will be verified by comparison with experimentally
obtained temperature dependent rate constants obtained in the Russell group. Together, these studies will provide molecular
level insight into the competing factors influencing cyclization.
An understanding of how these factors effect the rate of cyclization
will allow the development of more effective anticancer prodrug warheads –
enediynes that cyclize and cause cell death only under certain, controlled
conditions.
Recent Results
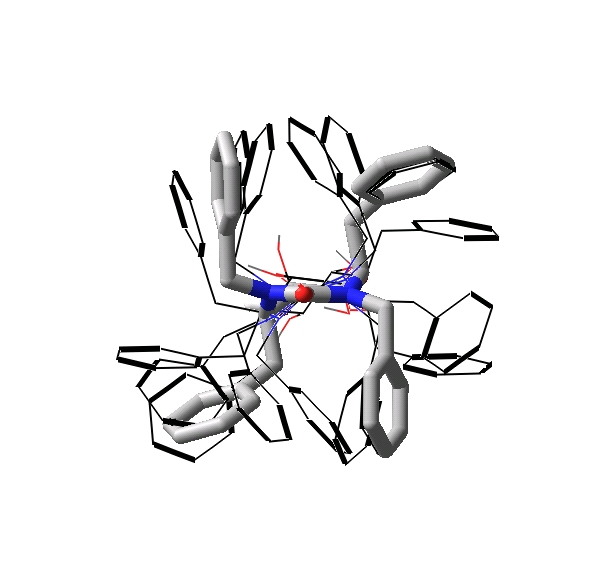 Comparing
the Conformational Behavior of a Series of Cyclic Urea HIV-1 Protease Inhibitors
The conformational flexibility of a series of diastereomeric cyclic
urea HIV–1 protease inhibitors has been examined using the Low Mode:Monte
Carlo conformational search method. (Journal
of Medicinal Chemistry, 2004, in press) An ensemble of low energy structures was generated
using OPLSAA/GBSA(water) and used to compare the molecular shape and flexibility
of each diastereomer to the experimentally determined binding affinities and
crystal structures of closely related systems. The results indicate that diastereomeric
solution-phase energetic stability, conformational rigidity and ability to
adopt a chair conformation correlate strongly with experimental binding affinities.
Rigid body docking suggests that all of the diastereomers adopt solution-phase
conformations suitable for alignment with the HIV-1 protease; however; these
results indicate that the binding affinities are dependent upon subtle differences
in the P1/P1' and P2/P2' substituent orientations.
Comparing
the Conformational Behavior of a Series of Cyclic Urea HIV-1 Protease Inhibitors
The conformational flexibility of a series of diastereomeric cyclic
urea HIV–1 protease inhibitors has been examined using the Low Mode:Monte
Carlo conformational search method. (Journal
of Medicinal Chemistry, 2004, in press) An ensemble of low energy structures was generated
using OPLSAA/GBSA(water) and used to compare the molecular shape and flexibility
of each diastereomer to the experimentally determined binding affinities and
crystal structures of closely related systems. The results indicate that diastereomeric
solution-phase energetic stability, conformational rigidity and ability to
adopt a chair conformation correlate strongly with experimental binding affinities.
Rigid body docking suggests that all of the diastereomers adopt solution-phase
conformations suitable for alignment with the HIV-1 protease; however; these
results indicate that the binding affinities are dependent upon subtle differences
in the P1/P1' and P2/P2' substituent orientations.
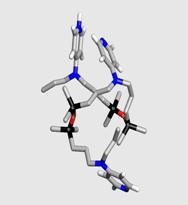 Analysis of Oligomeric Models of Synthetic, Silicon-based
Enzyme Catalysts: We
have investigated the conformational flexibility of hybrid organosiloxane
oligomers that contain aminopyridine groups in various states of protonation.
(Journal of Inorganic and Organometallic
Polymers, 2002 12(1-2), 31-47.) We are interested in these systems because
they are models for larger synthetic oligomers that display acyltransferases
enzyme-like properties. In this work,
an ensemble of low energy structures was generated and used to investigate
the dependence of molecular shape and flexibility on protonation. The results, as measured by the number of unique
conformations, the end-to-end or longest intramolecular distance and the radius
of gyration of the conformational point cloud, indicate that the number of
protonated pyridines plays a significant role in the overall molecular shape;
i.e., molecules with multiple pyridinium ions are, on average, significantly
larger, less flexible and more extended than molecules without charge.
These results help to explain the molecular behavior of the synthetic
catalytic systems and should guide future synthesis and molecular design of
synthetic enzymes.
Analysis of Oligomeric Models of Synthetic, Silicon-based
Enzyme Catalysts: We
have investigated the conformational flexibility of hybrid organosiloxane
oligomers that contain aminopyridine groups in various states of protonation.
(Journal of Inorganic and Organometallic
Polymers, 2002 12(1-2), 31-47.) We are interested in these systems because
they are models for larger synthetic oligomers that display acyltransferases
enzyme-like properties. In this work,
an ensemble of low energy structures was generated and used to investigate
the dependence of molecular shape and flexibility on protonation. The results, as measured by the number of unique
conformations, the end-to-end or longest intramolecular distance and the radius
of gyration of the conformational point cloud, indicate that the number of
protonated pyridines plays a significant role in the overall molecular shape;
i.e., molecules with multiple pyridinium ions are, on average, significantly
larger, less flexible and more extended than molecules without charge.
These results help to explain the molecular behavior of the synthetic
catalytic systems and should guide future synthesis and molecular design of
synthetic enzymes.
Optimizing
Conformational Search Methods:
We have recently completed the evaluation and comparison of two widely
used methods for searching the conformational space of complex molecules (Journal of Molecular Graphics and Modeling
2002, 21, 129-150). We
compared the ability of Low Mode,